![]()
![]()
BioPortainer Workbench is an open-source software developed under the MIT license and designed in a modular way, aimed at facilitating user interaction with Docker environments in three different computational layers: (i) infrastructure, (ii) platform and (iii) application. Its basic structure is briefly described in Figure 1. To deploy the software, user must initially access the BioPortainer Workbench image (Figure 1b) and install it, either in a single Docker engine, or in a Swarm cluster (Figure 1c). Once installed, BioPortainer Workbench (Figure 1d) consists of two basic containers: the BioPortainer Panel and the BioPortainer Pipeline Runner (Figure 1e). From a functional point of view (Figure 1f), the two containers offer a number of tools that allow users to perform a series of actions in the created Docker environmens, such as: (i) managing Docker resources associated with the BioPortainer Workbench; (ii) installing bioinformatics applications based on several platforms described in the literature and (iii) launching different types of analyses, using either command-line (CLI), or graphic-based interfaces (GUI). Such analyses may be conducted with the help of BioPortainer Workbench’s own resources (Figure 1g), or with resources harnessed from external repositories which provide preconfigured images, files, commands or scripts for the execution of bioinformatics software, with varying levels complexity (Figure 1h).
The BioPortainer Workbench image, as shown in Figure 1 (b), consists of a Docker Compose file, which is responsible for building the Docker environment on the host machine using Dockerfile files, which are associated with the two main containers of the software: the BioPortainer Panel and the BioPortainer Pipeline Runner. The third component of the image is a template repository, containing a series of JSON files, responsible for building the templates associate with platform installation, as well as for building the GUI forms used to launch bioinformatics tools with the aid of the BioPortainer GUI Runner.
The Docker Compose file contains all the necessary settings for complete system operation, including the execution settings of the two main containers (BioPortainer Panel and BioPortainer Pipeline Runner). In addition to these two main modules, there is a third module (not shown in Figure 1) called the BioPortainer Watchtower, designed to monitor the main containers during execution. This module also monitors the original BioPortainer Workbench images, updating the whole Docker environment, whenever new versions of the software are made available. The Docker Compose file can be easily edited with the aid of any text editor, so as to expand its functionality by incorporating additional features, such as web proxies and/or tools for Continuous Integration & Continuous Delivery (CI/CD), such as Jenkins, for example. To assist in the incorporation of these new resources, a virtual network, called BioPortainer Local, was created, in order to guarantee efficient communication between the containers. Thus, if users wish to incorporate new features into the BioPortainer Workbench, it is necessary to insert such features in this network, through the networks parameter, to guarantee their efficient integration to the different modules of the software.
The second component of the image are the Dockerfile files, which contain instructions for building the Docker images of the BioPortainer Panel and BioPortainer Pipeline Runner modules. These Dockerfiles are available through GitHub and can be easily expanded to accommodate additional needs of any user. The Dockerfile of the BioPortainer Panel module was developed from the original source file of Portainer, which has been modified to incorporate specific features of the BioPortainer Workbench, such as the tools BioPortainer JobRunner and BioPortainer GUI Runner. The Dockerfile of the BioPortainer Pipeline Runner, on the other hand, has been developed independently and presents a greater level of complexity in its structure, as it carries all the software and library prerequisites necessary for the execution of NextFlow scripts (Di Tommaso et al., 2017) in a Docker-in-Docker environment (DinD). In addition, given the characteristics of this complex environment, the BioPortainer Supervisor tool (not shown in Figure 1) has been added to this Dockerfile, providing users with a GUI (accessed through port 7000) that enables full management of the BioPortainer Pipeline Runner tool, the Jupyter Notebook and the Docker-in-Docker environment. It also allows users to analyze execution logs and control startup, shutdown and restart of processes within containers.
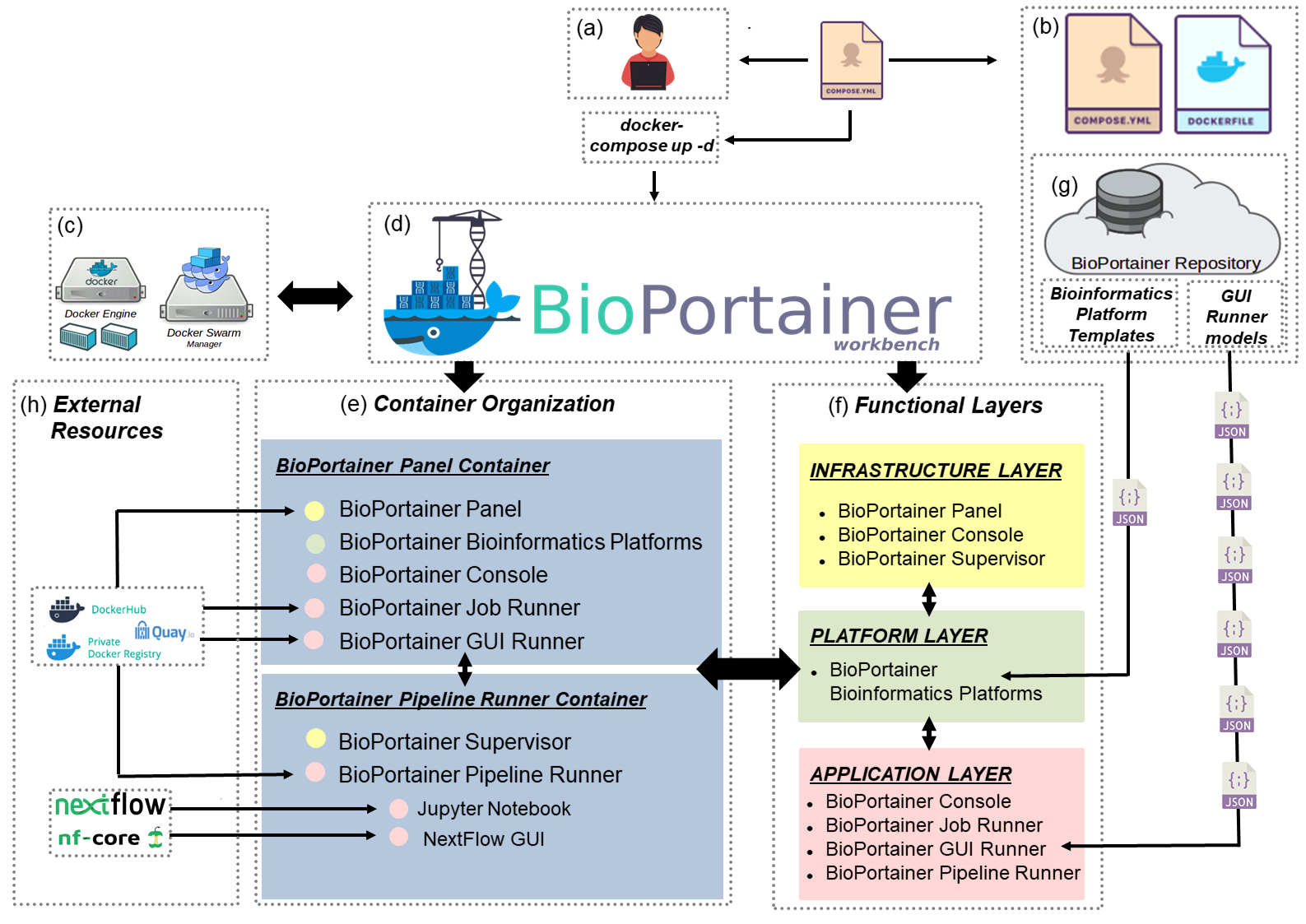
The third component of the image is the BioPortainer Repository, consisting of a series of JSON files. The JSON (JavaScript Object Notation) language allows storage of data structures in a standard interchange format, which can be used for transmitting data between a server and a graphical web interface application. One of these JSON files is responsible for generating the GUI templates that will assist users during the installation of the BioPortainer Bioinformatics Platforms (accessible though the BioPortainer Panel main menu), The BioPortainer Repository also carries additional JSON files that provide users with GUIs containing the interactive forms that assist in the launching of bioinformatics tools though the BioPortainer GUI Runner. All JSON files available at the BioPortainer Repository were manually developed to ensure perfect adaptation to the specific environment variables and parameter prerequisites of their specific platforms/tools. Moreover, all of them were individually tested by expert curators and further evaluated by Continuous Integration & Continuous Delivery (CI/CD), using the tools TravisCI and CircleCI. Users interested in modifying the BioPortainer Workbench JSON files, in order to expand their resources and/or adapt them to new needs, can obtain the individual files from the BioPortainer Workbench project webpage.
MENU
Installing and Configuring the BioPortainer Workbench [menu]
Installation of the BioPortainer Workbench is extremely simple and requires only Docker and Docker Compose as initial requirements. After both componets are installed, only two steps are needed to start a BioPortainer Workbench environment. In the first step, the compose.yml file is downloaded from the server (GitHub, or BioPortainer Workbench homepage) to the BioPortainer folder, in the host machine. In the second step, the Docker Compose is executed, downloading the images for the standard BioPortainer Workbench modules and the service is started. When Linux is the host machine’s operating system, the following commands must be run on the terminal:
Two steps are required to start a container containing BioPortainer:
$ wget https://goo.gl/bNecPA -O docker-compose.yml
$ docker-compose up -d
During the deployment process, some ports and disk volumes will be automatically configured in the host machine. Details on the ports and volumes created are available in the BioPortainer Workbench User Manual, which accompanies this manuscript as a supplementary file. In a standard implementation, the BioPortainer Workbench will use the localhost address (IP 0.0.0.0) as the default address for its internal links. If access is not performed through a local network, additional settings must be made in the Compose file, according to Docker’s official documentation, available here.
BioPortainer Workbench User Manual [menu]
For more information on different methods of installing and using the BioPortainer Workbench, visit the BioPortainer Workbench User Manual.
Author [menu]
Current development is led by Fabiano Menegidio.
Contributing [menu]
You are invited to contribute new features, fixes, or updates, large or small; we are always thrilled to receive pull requests, and do our best to process them as fast as we can.
Before you start to code, we recommend discussing your plans through a GitHub issue, especially for more ambitious contributions. This gives other contributors a chance to point you in the right direction, give you feedback on your design, and help you find out if someone else is working on the same thing.
Open Science Framework Repository [menu]
Identifier: DOI 10.17605/OSF.IO/2AZYG
MIT License [menu]
Permission is hereby granted, free of charge, to any person obtaining a copy of this software and associated documentation files (the “Software”), to deal in the Software without restriction, including without limitation the rights to use, copy, modify, merge, publish, distribute, sublicense, and/or sell copies of the Software, and to permit persons to whom the Software is furnished to do so, subject to the following conditions:
The above copyright notice and this permission notice shall be included in all copies or substantial portions of the Software.
The software is provided “as is”, without warranty of any kind, express or implied, including but not limited to the warranties of merchantability, fitness for a particular purpose and noninfringement. in no event shall the authors or copyright holders be liable for any claim, damages or other liability, whether in an action of contract, tort or otherwise, arising from, out of or in connection with the software or the use or other dealings in the software.
![]()
![]()